This contains links to genetics, statistics, and computer programs for linkage and association analysis, including my own. It was last updated on 09-Aug-2020.
David L. Duffy, MBBS PhD.
QIMR Berghofer Medical Research Institute,
300 Herston Road,
Herston, Queensland 4006, Australia.
Email: David.Duffy@qimrberghofer.edu.au.
Some photos from our Tasmanian holiday 2000.
Some photos from our UK holiday 2010.
Some photos showing climbing 2001-18.
Paintings.
| QIMR Berghofer and departmental links | My software | ||
| My CV/Publications | |||
| About asthma | |||
| About genetics | |||
| A genetic map | |||
| My links to other sites: genetics etc | |||
| Cycling at QIMR |
These are periodically updated reviews of the genetics of atopy and asthma. The articles include:
These chapters are based on my doctoral thesis, which is available in PDF format here.
Our publications on the genetics of allergic disease include:
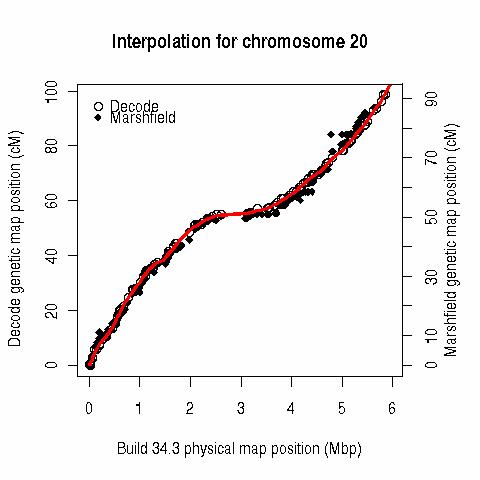
This table (last updated 20060618 18:40) contains interpolated genetic map positions for 128115 marker loci. The positions are in "Rutger's" cM (Kong X, Murphy K, Raj T, He C, White PS, Matise TC. A combined linkage-physical map of the human genome. Am J Hum Genet 2004; 75:1143-1148), estimated via locally weighted linear regression (lo(w)ess) from the Build 35.1 (and 34.3) physical map positions and published Rutgers genetic map positions ( R code here), and linearly interpolated "Oxstats" cM positions (Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 2005; 310: 321-324). A major difference between these two metrics is the model for recombination across the centromere.
For the pseudoautosomal region, I have interpolated a male map based on the sperm typing data of Lien et al [2000]. This is a separate file.
A research note describing this map is:
Duffy DL (2006). An integrated genetic map for linkage analysis.
Behavior Genetics 36: 4-6.
|
|
Duplicate markers have been removed. Beware of name modifications (eg Marshfield names often have letter code suffixes such as Z i.e. "Primer moved from its initial position; the allele size changed").
| 1 | Marker name |
| 2 | Alternative name |
| 3 | Alternative name 2 |
| 4 | Locus type (STS or SNP) |
| 5 | Decode marker (D or .) |
| 6 | Marshfield marker (M or .) |
| 7 | Chromosome |
| 8 | Alternative chromosome (occasionally differs!) |
| 9 | Decode order number (1-5136) |
| 10 | Marshfield order number (1-8010) |
| 11 | Decode physical position on chromosome (bp) |
| 12 | Decode genetic map position (cM) |
| 13 | Marshfield genetic map position (cM) |
| 14 | Rutger genetic map position (cM) |
| 15 | Rutger male genetic map position (cM) |
| 16 | Rutger female genetic map position (cM) |
| 17 | Build 34.3 physical map position (bp) |
| 17 | Build 35.1 physical map position (bp) |
| 18 | Interpolated physical map position (bp) |
| 19 | Interpolated Rutger genetic map position (cM) |
| 20 | Interpolated Oxstats genetic map position (cM) |
I have taken the chromosome band data used by NCBI Mapview to draw ideograms, and interpolated the band positions onto the above map (as opposed to the perhaps more logical approach of mapping linkage findings to a physical map!):

| SIB-PAIR |
I am making Fortran 95 source code and some binaries for my program SIB-PAIR available for downloading.
Sib-pair has had many bugs shaken out, though addition of newer commands leads to introduction of newer errors. While loading data could still be further improved for large datasets, analysis of data once in memory is fairly fast, so the program can be used for handling and analysis of genome-wide association study (GWAS) and smaller processed sequencing dataset. Since the May 2012 addition of multithreading (OpenMP), such analyses are now much faster. Very large datasets can also be analysed - data exceeding the available memory is automatically handled on disk rather than in memory (with a drop in speed).
The most recent version of Sib-pair (1.00b) is dated 8th August 2020 (see the list of new features). With respect to urgency of upgrading, for SIB-PAIR it is always a good idea! For example, map liftover using a chain file was not complete if the markers were unsorted - this was fixed 20180731. And the 20200221 changes include a nasty bug affecting merging of plink bed files. Note that the Windows binaries here may be a bit behind.
Program SIB-PAIR performs a number of analyses of family data that tend to be "nonparametric" or "robust" in nature. The name is a misnomer in that Sib-pair is actually for the analysis of arbitrary pedigrees. It is modelled to some extent on the Genetic Analysis System [Young, 1995] in terms of the command language and types of analysis. Included are routines for:
More recent releases of SIB-PAIR add multithreading, flexible manipulation of pedigree data, MLE of allele frequencies, segregation analysis, variance components (linkage) analyses that now allow multiple fixed effects including measured genotypes under Gaussian and threshold (probit-normal) models via deterministic approaches and a larger range of GLMMs via MCMC, the combined sibship/transmission disequilibrium score test for allelic association, an extension of the WQLS test to categorical traits, a quantitative trait TDT, the SKAT test, generalized linear (mixed) models, assorted classical twin analyses and one for bivariate survival analysis, multilocus population genetic analyses and estimation of empirical kinship coefficients.
The program executable is usually called sib-pair or sib-pair.exe. Precompiled executables are available for Linux and for Windows (see below), but there should be no problems compiling and running on platforms that have a Fortran 95 compiler. There are no hard coded constraints on number of loci, number of pedigree members or number of alleles at a marker (providing your computer has enough memory).
Using the japi library, a graphical file picker or directory browser is now working under Windows and Linux. An alternative uses the GTK2+ based pilib library. If these are not activated, there is a fallback simple text based file chooser.
The gfortran compiled code on linux is currently faster or as fast as any of the other compilers I use. Since some routines use formatted stream access, the program will not compile with some Fortran95 compilers.
Linux
MacOSX
This program generates nuclear families, a proportion of which contain monozygotic twins, in which multiple quantitative trait loci are segregating. One of these QTLs is linked to multiple markers. Families can be selected to contain high and/or low values at the quantitative or ordinal trait.
This is a Basic program that performs a number of simple statistical analyses of contingency tables useful in epidemiology and genetics. One can estimate tetrachoric correlations and odds ratios for 2x2 tables (with exact confidence intervals), combine multiple 2x2 tables via Mantel-Haenszel and maximum likelihood procedures (jackknife standard error for pooled MLE odds ratio), test for symmetry and quasi-symmetry in square contingency tables, and obtain exact (Pearson-Clopper) 95% confidence intervals on a proportion. A calculator (double precision) with scientific functions including inht(), fact(), and ran() is also accessible via the same menu.
rcexact. A program that calculates Fisher exact P-values for RxC contingency tables. Written by Mehta in Fortran 77 (Algorithm 643 from the ACM). I have altered the driving program slightly.
drawhap.sh. Takes SIMWALK2 haplotyping output file and draws the pedigree as a marriage-node graph with haplotypes using Graphviz (needs sh, awk, dot). Not completely satisfactory in terms of placement of haplotypes on the drawing. Colouring is of alleles, rather than haplotypes.
join_unsorted.sh. Just like (unix) join, but files do not have to be sorted. Returns a file following the order of the key in the first named file:
Usage: join.unsorted [OPTION]... FILE1 FILE2
For each pair of input lines with identical join fields, write a line to
standard output. The default join field is the first, delimited
by whitespace. When FILE1 or FILE2 (not both) is -, read standard input.
-a FILENUM print unpairable lines coming from file FILENUM, where
FILENUM is 1 or 2, corresponding to FILE1 or FILE2
-e EMPTY replace missing input fields with EMPTY
-i, --ignore-case ignore differences in case when comparing fields
-j FIELD equivalent to -1 FIELD -2 FIELD
-o FORMAT obey FORMAT while constructing output line
-t CHAR use CHAR as input and output field separator
-v FILENUM like -a FILENUM, but suppress joined output lines
-1 FIELD join on this FIELD of file 1
-2 FIELD join on this FIELD of file 2
--help display this help and exit
--version output version information and exit
Unless -t CHAR is given, leading blanks separate fields and are ignored,
else fields are separated by CHAR. Any FIELD is a field number counted
from 1. FORMAT is one or more comma or blank separated specifications,
each being FILENUM.FIELD or 0. Default FORMAT outputs the join field,
the remaining fields from FILE1, the remaining fields from FILE2, all
separated by CHAR.
fscheme. Port of the tinyscheme (and minischeme) small Scheme interpreter to Fortran 95. Hopefully useful as an embedded interpreter (a stripped down version is present as a module in Sib-pair).
grapheps. Port of Aubrey Jaffer's grapheps Postscript data plotting package to a Fortran 95 module. As used in Sib-pair.
fortransockets. Minimal Fortran 95 sockets library for linux. Enough functionality for a simple server. Includes wrappers for socket(), setsockopt(), bind() and listen(); accept(); send(); recv(); close(); gethostbyname(). This code last updated 2007-12-19.
Sample code for multidimensional table. Enhanced version of Sib-pair code for sorting and tabulating multidimensional data. For my purposes, the inputs to set_table_cell would be character data, which would then be automatically cast to the correct type.
Interface for curses. Fortran 2003 modules that interface to the PDCurses and ncurses libraries. With PDCurses, this runs nicely using gfortran or g95 on linux or Windows. The ncurses and PDCurses libraries differ in a few places in their coding for attributes and some keys (eg backspace). This code last updated 2011-02-01.
A couple of awk scripts used to make the interfaces to C (usually will need some editing afterwards).
Interface to zlib. After Janus Weils's example on comp.lang.fortran (May 2009) and fgzlib (plus templates in fgsl). As others have noted, it is much faster to use gzread() and a buffer, rather than gzgets(). Extended slightly (2018-10-02), to interface the necessary routines to randomly access a BGZF (ie bgzipped) file.
Interface to readdir() etc. A module that allows listing directory contents using the Posix opendir, readdir, rewinddir, closedir. Works on Linux and Darwin.
David Frank's C2F translator. This was available for many years at his website, but this seems defunct. He wrote about it on comp.lang.pl1 back in 2002 that:
I made my C2F tool freeware from its beginning several years back, BTW, I'm a old retired Fortran programmer, but if I were to go back into the job market I would certainly add this project to my resume,,,
JabRef reference formatting style files for assorted biomedical journals. The layout files are used to define a customized export format for a set of references. In this case, they produce a character delimited file which after postprocessing to make it nicely tab separated, can be uploaded to the Australian NHMRC grant application system (RGMS).