Data on pedigree structure and the phenotypes of interest are collected by interviewers/field workers and usually entered into databases using the program CYRILLIC. This program allows automated drawing of pedigrees as well as acting as a front end for a sophisticated database.
Other programs are now preferred (I believe that Cyrillic is no longer supported). We have tried Progeny, which offers a complete databasing/drawing environment, but this does not interact well with Oracle, where all our existing data is kept.
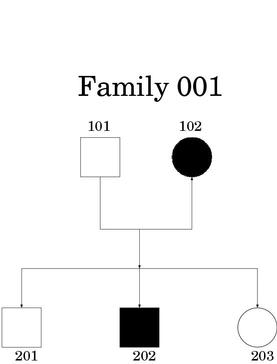
We use CYRILLIC therefore mainly to store the pedigree structure as it is much easier to check pedigree details with a well drawn pedigree diagram to hand. Oracle databases also retain the parental IDs for each individual. This information is then exported in the GEDCOM format -- a general purpose genealogical form and converted by shell scripts into the standard LINKAGE or GAS format eg :
From:
To:
| Pedigree_ID | Individual_ID | Father_ID | Mother_ID | Sex | Phenotype1 |
|---|---|---|---|---|---|
| 001 | 0001101 | x | x | m | n |
| 001 | 0001102 | x | x | f | y |
| 001 | 0001201 | 0001101 | 0001102 | m | n |
| 001 | 0001202 | 0001101 | 0001102 | m | y |
| 001 | 0001203 | 0001101 | 0001102 | f | n |
The GAS format is a simple text file containing one line for each family member. The first five columns (fields) are compulsory and contain the family and individual IDs the parental IDs and the sex. The individual and parental IDs are the minimum sufficient information to be able to reconstruct the pedigree successfully. Founders (individuals whose parents are not included in the pedigree drawing) have x, the missing value marker, in their parent fields. For a pedigree to be technically correct, all individuals must either have zero or two parents recorded in these fields. However, many computer programs will automatically generate a dummy parent to include in the pedigree file if only one parent has been specified.
In a standard pedigree drawing the phenotype at a binary trait (yes or no affected or unaffected) is displayed by filling the square or circle. This has been recorded in the 6th field of the pedigree file as y or n. Additional fields containing quantititive data such as age, or genotype data, can also be present.
The genotype at a Simple Sequence Repeat (SSR) marker is expressed as the length of each of the two alleles in base pairs. Formerly, the usual program used at QIMR to score migration of fluorescently labelled DNA on electrophoretic gels was Genotyper. The output after scoring an individual at a number of markers is the best estimate of the allele length corresponding to the peak fluorescence for each allele.
| Unique ID | Marker | Length Allele 1 | Length Allele 2 | Gel |
|---|---|---|---|---|
| 0001101 | D3S3000 | 265.63 | 277.20 | SYS2-Z-1 |
| 0001101 | D3S2000 | 130.78 | 134.80 | SYS2-Z-1 |
| 0001101 | D3S1500 | 282.08 | 313.26 | SYS2-Z-1 |
| 0001101 | GATA333E0 | 149.66 | 157.79 | SYS2-Z-1 |
| 0001102 | D3S3000 | 265.84 | 269.81 | SYS2-Z-1 |
| 0001102 | D3S2000 | 130.93 | 134.96 | SYS2-Z-1 |
| 0001102 | D3S1500 | 278.37 | 313.58 | SYS2-Z-1 |
| 0001102 | GATA333E0 | 149.65 | 149.65 | SYS2-Z-1 |
| 0001201 | D3S3000 | 265.84 | 269.61 | SYS2-Z-1 |
| 0001201 | D3S2000 | 130.93 | 134.96 | SYS2-Z-1 |
| 0001201 | D3S1500 | 278.37 | 313.58 | SYS2-Z-1 |
| 0001201 | GATA333E0 | 149.65 | 157.30 | SYS2-Z-1 |
| 0001202 | D3S3000 | 265.84 | 269.81 | SYS2-Z-1 |
| 0001202 | D3S2000 | 130.41 | 134.66 | SYS2-Z-1 |
| 0001202 | D3S1500 | 278.31 | 313.58 | SYS2-Z-1 |
| 0001202 | GATA333E0 | 149.98 | 149.65 | SYS2-Z-1 |
| 0001203 | D3S3000 | 265.84 | 269.81 | SYS2-Z-1 |
| 0001203 | D3S2000 | 130.93 | 135.25 | SYS2-Z-1 |
| 0001203 | D3S1500 | 278.86 | 313.63 | SYS2-Z-1 |
| 0001203 | GATA333E0 | 149.65 | 149.77 | SYS2-Z-1 |
Under UNIX, these genotypes can be combined with the pedigree structure described by the GAS file using standard utilities such as sort, awk and join. This Bourne shell script performs this action.
Alternatively, this can be performed within any standard database or spreadsheet. Eventually we obtain a file like the following:
| Pedigree_ID | Individual_ID | Father_ID | Mother_ID | Sex | Phenotype1 | D3S3000 | D3S2000 | D3S1500 | GATA333E0 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 001 | 0001101 | x | x | m | n | 265.63 | 277.20 | 130.78 | 134.80 | 282.08 | 313.26 | 149.66 | 157.79 |
| 001 | 0001102 | x | x | f | y | 265.84 | 269.81 | 130.93 | 134.96 | 278.37 | 313.58 | 149.65 | 149.65 |
| 001 | 0001201 | 0001101 | 0001102 | m | n | 265.84 | 269.61 | 130.93 | 134.96 | 278.37 | 313.58 | 149.65 | 157.30 |
| 001 | 0001202 | 0001101 | 0001102 | m | y | 265.84 | 269.81 | 130.41 | 134.66 | 278.31 | 313.58 | 149.98 | 149.65 |
| 001 | 0001203 | 0001101 | 0001102 | f | n | 265.84 | 269.81 | 130.93 | 135.25 | 278.86 | 313.63 | 149.65 | 149.77 |
Binning refers to replacing the approximate allele lengths above with the most likely "true" length, given that the length will actually be an integer number of base pairs. Furthermore, most SSRs, as the name implies, will exist in different forms (allelomorphs or alleles) that are an integer number of repeats of a short sequence of base pairs, usually 2, 3 or 4. To confuse matters, there are "tetra" nucleotide repeat markers with a few interspersed "half" alleles (generated by a different mutational mechanism). Standard databases such as GDB will give population results for a particular marker.
The most usual microsatellite genotype data we see now is from commercial standard sets of markers for genome scans. All the assays are optimized, and the alleles sizes well known. This makes the binning a lot simpler.
The actual methods use to genotype SNP markers are changing rapidly. For linkage scans and for association scans of the genome or even particular chromosomal regions, again there are now standard sets from different manufacturers. The linkage SNP panels (3600, 10000 SNP) will soon replace STRPs for linkage genome scans because of their more even coverage.
We deal with data from the Sequenom MassARRAY system, from Affymetrix and Illumina SNP chips, Because of cost reasons (with the new multiplexing assays, approximately 1 cent per genotype) the Sequenom genotypes are usually for candidate regions. The 100K and upward SNP chips represent a lot of data for analysis, but in theory the processes are much the same. The Affymetrix genotyping calling software has previously been suboptimal, but recent work by Terry Speed and others has greatly improved this. In our hands, the Illumina results are extremely high quality in terms of call rate and genotyping error.
Before proceeding to analysis of the chosen phenotype, we usually perform further tests for genotyping errors. One of these is to perform multipoint linkage analysis of the markers. If markers are tightly linked, then the probability of two close recombination events is quite small. Broman and Weber [2000] suggest the probability of a double recombinant within a 20 cM interval is only 2 in 1000. If one observes such a tight double recombinant within human family data, then it is 5-10 times more likely that this represents a genotyping error at a marker separating the two recombination events.
This type of procedure is automated in freely available programs, notably MERLIN (--errors) and MENDEL.
There are several mechanisms that can generate systematic errors in a pedigree. These include deliberate or accidental errors in an informant's description of a family, nonpaternity, and sample mix-up. There are several computer programs that can assess the degree of relationship between individuals based on their genotype at multiple markers.
We use both RELATIVE and RELPAIR. SIB-PAIR can used to produce the correct pedigree files for both these programs. For data that deals with twins, RELPAIR is the better program, as it can detect undiagnosed monozygotic twins. RELPAIR also checks across all individuals versus all individuals in the file, testing for sample mixups across pedigrees.
The programs GENEHUNTER and ALLEGRO both offer rapid estimation of haplotypes in small to medium sized pedigrees. These can then be used to asses the number and location of crossovers. These programs require the pedigree file to be in the LINKAGE format, and also need a locus file describing the pedigree file.
We often use the program SIB-PAIR to prepare these files. SIB-PAIR uses a similar control file to that required by BINNING. To automatically generate a linkage map for the LINKAGE format locus file, it is necessary to sort the pedigree marker genotype fields into map order.
We first create a SIB-PAIR control file that is in the correct map order:
set locus trait aff
set locus D3S1500 marker 56.3 centiMorgans
set locus D3S3000 marker 59.1 centiMorgans
set locus GATA333E0 marker 59.8 centiMorgans
set locus D3S2000 marker 70.1 centiMorgans
read pedigree ordered01.ped
run
write locus linkage ordered01.loc
write linkage ordered01.pre
One way to prepare such a file is to use the shell script mastermap, which searches the LDB files for the position of the included markers, so:
davidD@orpheus> mastermap bin01.in ordered01.in
You can simply type mastermap to get help on running this script.
Once this file is prepared (called ordered01.in, say), we can use another UNIX shell script to reorder the columns of the pedigree file:
davidD@orpheus> reorder bin01.in bin01.ped ordered01.in ordered01.ped
davidD@orpheus> sib-pair < ordered01.in > ordered01.out
or one can reorder once the pedigree file has been read in by most genetic analysis programs, including Sib-pair.
We have now created the appropriate LINKAGE style locus and pedigree files. To use ALLEGRO to produce haplotypes, we make an ALLEGRO type control file, containing the following commands:
datfile ordered01.loc
prefile ordered01.pre
haplotype
crossoverrate
This file (called ordered1.opt, say) is then used in the following command:
davidD@orpheus> allegro ordered01.opt
ALLEGRO will write several output files in response to these commands. The files inher.out, haplo.dat and ihaplo.dat contain the haplotyping information, while xover.dat contains the estimated recombination rates between each pair of adjacent markers.
It is often not fruitful to spend too much time on following up isolated genotyping errors, but a pattern of errors in one family or with one marker can point to severe problems affecting the entire analysis. See the accompanying document for the effects of errors on analysis.